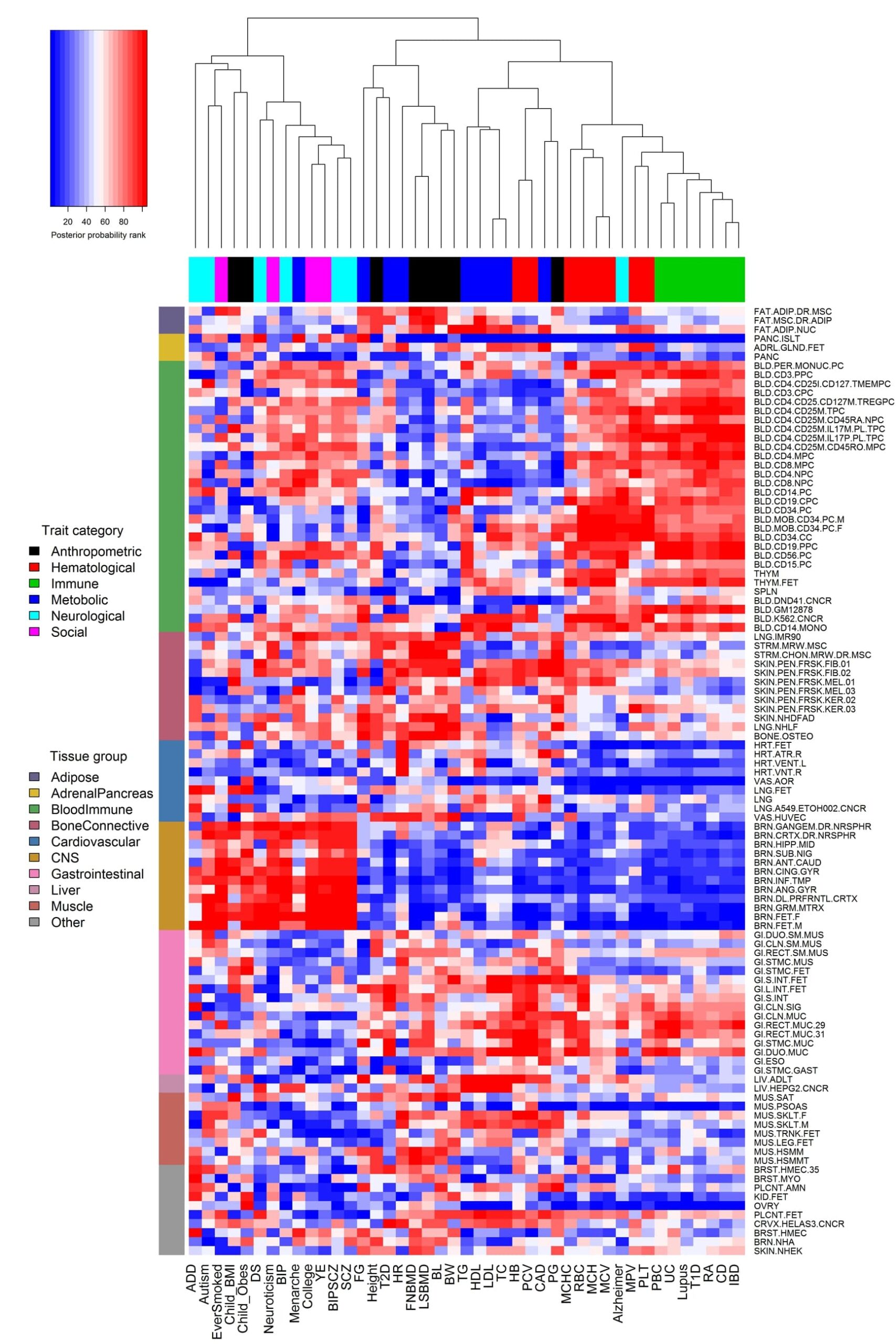
My research is focused on developing efficient and effective statistical and computational methods for genetic and genomic studies. These studies often involve large-scale and high-dimensional data; examples include genome-wide association studies, epigenome-wide association studies, and various functional genomic sequencing studies such as bulk and single cell RNAseq, bisulfite sequencing, ChIPseq, ATACseq etc. Our method development is often application oriented and specifically targeted for practical applications of these large-scale genetic and genomic studies, thus is not restricted in a particular methodology area. Our previous and current methods include, but are not limited to, Bayesian methods, mixed effects models, factor analysis models, sparse regression models, deep learning algorithms, clustering algorithms, integrative methods, spatial statistics, and efficient computational algorithms. By developing novel analytic methods, I seek to extract important information from these data and to advance our understanding of the genetic basis of phenotypic variation for various human diseases and disease related quantitative traits.